| |
КОТЛЫ ПАРОВЫЕ
НИЗКОГО И СРЕДНЕГО ДАВЛЕНИЯ.
ОРГАНИЗАЦИЯ И МЕТОДЫ ХИМИЧЕСКОГО КОНТРОЛЯ
ЗА ВОДНО-ХИМИЧЕСКИМ РЕЖИМОМ
(РТМ 24.030.24-72)
Настоящий РТМ составлен из расчета использования его
указаний в промышленных или отопительных котельных, комплектуемых барабанными
котлами низкого и среднего давления по ГОСТ 3619-69, изготовляемыми заводами
Минтяжмаша для давления 0,9 - 4,0 МПа (9 - 40 кгс/см2) и
паропроизводительности от 0,25 до 75 т/ч.
РТМ может быть
распространен и на котлы соответствующей паропроизводительности и параметров,
изготовленные заводами других ведомств, а также на импортные котлы при условии
получения соответствующего подтверждения ведомственной наладочной
специализированной организации.
РТМ определяет
требования по организации химического контроля, предъявляемые к
заводам-изготовителям котлов, проектирующим организациям и предприятиям,
осуществляющим эксплуатацию котлов.
СОДЕРЖАНИЕ
1.
ЗАДАЧИ И ОБЪЕМ ХИМИЧЕСКОГО КОНТРОЛЯ
1.1.
Химический контроль за качеством воды и пара в промышленных котельных должен
обеспечить надежную и экономичную эксплуатацию всех аппаратов и элементов
тепловой схемы энергетической установки и в первую очередь самих котельных
агрегатов.
Важной задачей
организации химического контроля является его автоматизация, унификация и
создание условий, которые позволили бы свести к минимуму трудоемкость и
количество выполняемых операций.
1.2.
Химический контроль осуществляется производством текущего оперативного контроля
за всеми стадиями водоподготовки, водно-химического режима котлов и теплообменных
аппаратов, а также углубленного периодического контроля за всеми типами вод -
от исходной до конденсата пара - с целью фиксации фактического режима
энергетической установки в целом.
1.3. Текущий
оперативный контроль должен производиться непрерывно при помощи автоматических
или полуавтоматических приборов и дополняться производством простых
приближенных аналитических определений.
При отсутствии
приборов для непрерывной регистрации показателей качества добавочной химически
обработанной и питательной воды рекомендуется для котельных всех типов
организовать отбор представительных среднесуточных проб этих вод с
производством в дневную смену их анализа.
Углубленный
периодический контроль должен давать четкое количественное представление о
составе исходной воды и динамике изменений этого состава в тракте котельной и в
системе водоподготовки во времени, о качестве конденсата, возвращаемого из
каждого теплообменного аппарата в питательную систему котлов, а также в
качестве пара, выдаваемого котлами.
1.4. Данные анализов,
в том числе и среднесуточных проб, должны давать возможность проведения
расчетов величины продувки котлов, влажности пара, величины возврата конденсата
в питательную систему котлов, эффективности работы обескислороживающей
установки.
Данные анализа
периодического контроля необходимы для установления основных показателей работы
водоподготовительной установки: удельного расхода реагентов, их дозы и
качества, емкости поглощения катионитов, грязеемкости фильтрующих материалов,
глубины освобождения воды от отдельных примесей и т.п. По данным анализов
ведутся также процессы коррекционной обработки воды: фосфатирование,
нитратирование и аминирование воды.
Результаты
анализов по определению содержания продуктов коррозии служат основанием для
оценки интенсивности протекания процессов повреждения металла в пароводяном
тракте энергетической установки.
1.5.
Необходимый объем контроля в каждой конкретной котельной определяется
конструктивными особенностями котлов, особенностями общей тепловой схемы,
принятым способом водоподготовки, качеством возвращаемого конденсата с учетом
технологических особенностей производства, степенью оснащенности приборами
химического контроля и автоматизацией процессов технологической схемы
водоподготовки.
Общий объем
контроля энергетической установки устанавливает с учетом настоящего РТМ
ведомственная или привлеченная наладочная организация.
1.6.
Рекомендуемый объем химического контроля за энергетическими установками,
работающими в условиях нормальной эксплуатации, указан в табл. 1.
(Буквой Н в табл.
1 обозначено, что рекомендуемые аналитические определения делаются
один раз в неделю.)
В пусковой и
наладочный период химический контроль устанавливает наладочная организация, при
этом объем его не должен быть меньше объема, предусмотренного настоящим РТМ.
1.7. Кроме
анализов воды и пара, в практике эксплуатации энергетических установок нередко
возникает необходимость анализа различного рода отложений для установления
причин их образования.
Такие
определения, так же как и полный анализ воды, обычно выполняет центральная
заводская лаборатория предприятия или водные лаборатории специальных
институтов, организаций и химических служб энергосистем Минэнерго.
2.1. Требования к заводам-изготовителям котлов.
2.1.1.
Устройства для отбора проб насыщенного пара питательной и котловой воды
изготовляют в соответствии с отраслевой нормалью ОН 24-3-69-66.
Устройства для
отбора проб перегретого пара в объем химического контроля не входят.
2.1.2. Каждый
отбор пробы в соответствии с ОН 24-3-69-66 оборудуется своим трубопроводом из
трубы диаметром не более Dу10, на котором устройства для
отбора проб располагаются в следующей последовательности:
зонд для
получения представительной пробы (пробоотборник);
запорный
вентиль Dу6, установленный за пробоотборником (для котлов
группы III устанавливают последовательно два вентиля);
холодильник,
установленный на водном щите;
дроссельный
игольчатый вентиль Dу6, установленный на выходе из
холодильника;
водный щит
(для всех отборных точек котельной).
Запорный и
регулирующий вентили Dу6 для загрязненных вод
допускается заменять на Dу10.
2.1.3.
Устройства для отбора проб необходимо изготовлять по чертежам, выполненным с
учетом требований настоящего руководящего технического материала и отраслевых
стандартов, изложенных в табл. 2.
2.1.4. Котлы
группы I (по табл.
1) оснащают устройствами для отбора проб питательной и котловой
воды, а котлы групп II и III - дополнительно устройствами для отбора проб
насыщенного пара. Устройства для отбора пробы питательной воды и котловой воды
на линии непрерывной продувки устанавливают при монтаже в соответствии с
проектом.
2.1.5. На
котлах группы II с пароперегревателем и на котлах группы III для отбора проб
насыщенного пара устанавливают устьевой зонд, а на котлах без пароперегревателя
- щелевой зонд.
Таблица 1
|
Тип котлов
|
Группа котлов
|
Возможные способы и фазы
водоподготовки 8
|
Вид воды
|
Рекомендуемые аналитические
определения
|
Рекомен-
дуемая категория лабора-
торий
|
|
Прозрач-
ность
|
Щелоч-
ность
|
Жесткость
|
Хлориды
|
Солесодер-
жание
|
Кислород
|
Фосфаты
|
Нитраты
|
Угле-
кислота
|
Аммиак
|
Железо
|
Медь
|
рН
|
Нитраты
|
Окисля-
емость
|
Масло
|
|
Число определений в сутки
|
|
Е-0,25-9 - Е-1,6-9 (ММЗ и
МКЗ)
|
I
|
Na-катионирование
|
Исходная
|
Н
|
Н
|
Н
|
Н
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
Первая
|
|
Термическая деаэрация
|
Химически обработанная
|
11
|
11
|
2(1)1
|
11
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Реагентная или
безреагентная обработка воды (внутрикотловая) 9
|
Конденсат
|
1
|
1
|
1
|
1
|
-
|
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Питательная
|
11
|
11
|
2(1)1
|
11
|
-
|
12
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Котловая
|
1
|
1
|
-
|
1
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Е-4,5-14 - Е-50-14; Е-4-14-225
- Е-20-24-250 (ДКВр)
|
II
|
Фильтрование, Na-катионирование, термическая деаэрация, аминирование
|
Исходная
|
1
|
1
|
1
|
1
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
Вторая
|
|
Осветленная
|
2
|
-
|
-
|
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Химически обработанная
|
11
|
11
|
2(1)1
|
13
|
11
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Конденсат
|
1
|
1
|
1
|
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
То же, с предварительной
коагуляцией
|
Питательная
|
11
|
11
|
2(1)1
|
13
|
11
|
1
|
|
|
1
|
1
|
Н6
|
-
|
1
|
-7
|
-7
|
-7
|
|
Котловая
|
1
|
2
|
-
|
23
|
2
|
-
|
25
|
25
|
|
|
|
|
|
|
|
|
|
Насыщенный пар
|
-
|
13
|
-
|
-
|
14
|
|
|
|
Н
|
|
|
|
|
|
|
|
|
Е-20-40 - Е-75-40 (котлы
БелКЗ)
|
III
|
Известкование,
фильтрование, Na-катионирование в две
ступени, термическая деаэрация, фосфатирование и аминирование
|
Исходная
|
1
|
1
|
1
|
1
|
-
|
-
|
-
|
-
|
-
|
-
|
Н
|
-
|
-
|
-
|
-
|
-
|
Третья
|
|
Осветленная
|
6
|
3
|
3
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
Н
|
-
|
-
|
-
|
-
|
-
|
|
Химически очищенная
|
3 (1)1
|
11
|
12 (1)1 3
|
-
|
11
|
-
|
-
|
-
|
-
|
-
|
Н
|
Н7
|
-
|
-
|
-
|
-
|
|
Конденсат
|
3
|
3
|
12(1)1
|
-
|
3
|
-
|
-
|
-
|
-
|
Н
|
Н
|
Н7
|
1
|
-
|
-
|
-
|
|
Питательная
|
3 (1)1
|
3 (1)1
|
-
|
13
|
3 (1)1
|
3
|
-
|
-
|
1
|
3
|
Н
|
Н7
|
1
|
-7
|
-7
|
-7
|
|
Коагуляция, фильтрование,
Nа-катионирование, аминирование, термическая деаэрация, гексаметафосфатирование
|
Котловая чистого отсека
|
|
3
|
-
|
-
|
3
|
-
|
65
|
65
|
-
|
-
|
Н
|
-
|
-
|
-
|
-
|
-
|
Третья
|
|
Котловая солевого отсека
|
3
|
6
|
-
|
-
|
3
|
-
|
Н 5
|
Н 5
|
-
|
-
|
Н
|
-
|
-
|
-
|
-
|
-
|
|
Насыщенный пар
|
-
|
33
|
-
|
-
|
-
|
-
|
-
|
|
Н
|
3
|
-
|
-
|
1
|
-
|
-
|
-
|
|
Подпиточная
|
3
|
3
|
3
|
-
|
-
|
1
|
-
|
-
|
-
|
-
|
Н
|
-
|
1
|
-
|
-
|
-
|
|
Сетевая
|
3
|
3
|
3
|
-
|
-
|
1
|
-
|
-
|
-
|
-
|
Н
|
-
|
1
|
-
|
-
|
-
|
|
|
Для всех групп
|
При использовании растворов
|
Растворы реагентов
|
Проверять концентрацию при приготовлении
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 Анализы производят только в
среднесуточных пробах. При обозначениях 2 (1)1 или 3 (1)1
цифра вне скобок указывает общее число отборов, а в скобках - число анализов из
среднесуточной пробы.
2 Только при наличии
обескислороживания питательной воды.
3 Анализ производят в случае
отсутствия определения солесодержания.
4 Только для котлов, имеющих
пароперегреватель.
5 В случае отсутствия
какой-либо из фаз обработки воды (фосфатирование, нитратирование)
соответствующий показатель качества воды не контролируют.
6 Только для котлов,
работающих на жидком топливе.
7 Анализ выполняют
эпизодически, только при наличии в исходной воде или, при необходимости, по
заключению наладочной организации.
8 При других способах
докотловой водоподготовки объем контроля соответственно корректирует
специализированная наладочная организация.
9 Замена докотловой обработки
воды, внутрикотловой (реагентной или безреагентной), а также необходимый
химический контроль допускаются и устанавливаются соответствующим заключением
специализированной организации. Н - один раз в неделю.
Таблица 2
|
Наименование устройства
|
Характеристика, номер
стандарта
|
Место установки
|
Требования к установке
|
|
Пробоотборные устройства:
питательной воды
|
Г-образная трубка.
ОН 24-3-70-66
|
Трубопровод питательной воды
|
Наличие прямого участка трубопровода длиною не менее 10 диаметров до
места установки и не менее пяти диаметров после него
|
|
котловой воды чистого отсека
|
|
Нижний участок опускной трубы первой ступени испарения
|
То же
|
|
котловой воды котлов без ступенчатого испарения и второй ступени
котлов со ступенчатым испарением
|
Приваренный штуцер
|
Линия непрерывной продувки котла
|
Приваривается штуцер заподлицо с внутренней поверхностью трубы.
Устанавливается до узла регулирования продувки
|
|
насыщенного пара котлов с пароперегревателем
|
Устьевой зонд.
ОН 24-3-85-66
|
Пароотводящая труба
|
Устанавливается в устье трубы для отвода пара из барабана таким
образом, чтобы выходное сечение наконечника располагалось в плоскости
входного сечения трубы
|
|
насыщенного пара котлов без пароперегревателя
|
Щелевой зонд
|
Паропровод насыщенного пара
|
Устанавливается за измерительной шайбой на расстоянии пяти диаметров
от диафрагмы
|
|
насыщенного пара из выносного цикла
|
Щелевой зонд.
ОН 24-3-40-66
|
На выходе из выносного циклона
|
Используется только в период пусконаладочных работ
|
|
Холодильники
|
Одноточечные 1 и двухточечные
|
Водный щит
|
-
|
|
Водный щит
|
ОН 24-3-97-66
ОН 24-3-118-77
|
По проекту
|
Характеризуется числом установок холодильников
|
1 Изготавливает Саратовский
завод тяжелого машиностроения.
На
котлах типа ДКВр с пароперегревателем пробоотборник устанавливают на первой к
фронту трубе пароперегревателя котла, а на котлах группы III - на двух
пароотводящих трубах с левой и правой сторон котла.
2.1.6.
Расчетная скорость отбираемой среды во входном отверстии устьевого зонда должна
быть такой же, как и в трубопроводе.
2.1.7.
Устройства для отбора проб котловой воды из котлов без ступенчатого испарения
устанавливают на линии непрерывной продувки котла. Пробы воды на котлах со
ступенчатым испарением для солевых отсеков отбирают из линии непрерывной
продувки каждого отсека, а для чистого отсека - из нижнего участка одной из опускных
труб этого отсека.
2.1.8. Для
предотвращения возможности перекоса химического состава котловой воды по длине
чистого отсека и вместе с тем для отбора средней представительной пробы в
случае необходимости в конструкции опускной системы этого отсека котла должно
быть предусмотрено соответствующее перекрещивание опускных труб.
2.1.9.
Змеевики холодильников для котлов группы I, а также для котловой воды котлов
групп II и III выполняют из стали 20.
Змеевики
холодильников для питательной воды и пара котлов групп I и II выполняют из
стали Х18Н12Т.
2.1.10. Для
котлов групп II и III, сжигающих мазут, для проб питательной воды и пара
применяют вентили запорные из коррозионно-стойкой стали (в целях получения
представительной пробы по содержанию соединений железа), а для котловой воды -
из углеродистой стали.
2.2. Требования к
проектирующим организациям.
2.2.1. В
зависимости от типа котельных устанавливают объем химического контроля в целом
по котельной в соответствии с табл. 1, а также выбирают место для организации
отбора пробы питательной воды на трубопроводе за питательным насосом и котловой
воды на линии непрерывной продувки котла.
2.2.2.
Проектируют в целом для всей котельной линии отбора проб, которые сводят на водный
щит, способствующий обеспечению удобства и представительности отбора проб.
Выбирают место для центрального общекотельного водного щита или нескольких
щитов в месте, удобном для обслуживания, в зависимости от местных условий
компоновки. К щиту проектируют подвод охлаждающей воды и организуют отвод от
него использованной воды и раздельный отвод чистых вод, используемых в
дальнейшем для питания котлов. Для осуществления обратных продувок
пробоотборных линий при их закупорке рекомендуется предусмотреть наличие
гидропресса.
2.2.3. В
соответствии с установленным объемом химического контроля составляют
спецификацию на поставку трубопроводов.
2.2.4.
Трубопроводы отбора всех проб для котлов группы I, а также для котловой воды
котлов групп I и II выполняют из стали 20.
Все остальные
трубопроводы отбора проб для котлов групп II и III выполняют из стали Х18Н12Т.
2.2.5.
Использование одного холодильника для отбора нескольких проб допускается только
при отборе проб насыщенного пара на котлах без пароперегревателя и на бойлерных
установках для контроля за качеством конденсата из каждого бойлера. Для отбора
проб котловой воды использовать один холодильник на несколько точек не
разрешается.
2.2.6. В
зависимости от типа котельных проектирующие организации предусматривают организацию
водной лаборатории трех категорий в соответствии с табл. 1.
2.2.7. Для
водной лаборатории первой категории специального помещения не
предусматривается. В котельных аналитический стол должен находиться в
застекленном блоке-кабине размером 6-8 м2.
Все
предусмотренные табл.
1 определения для котлов группы I, кроме определения прозрачности,
рекомендуется производить, используя экспресс-лабораторию анализа воды (ЭЛВК-5)
и кислородомер типа ОКВ. Все необходимые реактивы для проведения анализов
готовят в центральной лаборатории предприятия или в специальных лабораториях
сторонних организаций.
Таблица 3
|
Наименование оборудования
|
Категория лабораторий
|
|
Первая
|
Вторая
|
Третья
|
|
Стол титрованных растворов
|
1
|
1
|
1
|
|
Холодильник для конденсации пара или
дистилляционный аппарат
|
-
|
1
|
1
|
|
Экспресс-лаборатория типа ЭЛВК-5
|
2
|
-
|
-
|
|
Лабораторная обессоливающая установка
|
-
|
1
|
1
|
|
Лабораторный солемер или мегомметр на 500 В
|
-
|
2
|
2
|
|
Электроплитки
|
-
|
2
|
2
|
|
Сушильный шкаф
|
-
|
-
|
2
|
|
Муфельная печь
|
-
|
-
|
2
|
|
Аналитические весы
|
-
|
-
|
1
|
|
Полуавтоматический анализатор кислорода ОКВ
|
1
|
2
|
2
|
|
Технические весы
|
-
|
1
|
1
|
|
Лабораторные катионитные фильтры
|
-
|
2
|
2
|
|
Вытяжной шкаф
|
-
|
-
|
1
|
|
Стол для нагревательных приборов
|
-
|
-
|
1
|
|
Шкаф для посуды и реактивов
|
-
|
1
|
1
|
|
Стол для приборов
|
-
|
1
|
1
|
|
Прибор для определения прозрачности
|
1
|
1
|
1
|
|
Стол лабораторный
|
-
|
1
|
2
|
|
Стол для аналитических весов
|
-
|
-
|
1
|
|
Лабораторные табуретки
|
-
|
2
|
3
|
2.2.8. Для водной лаборатории второй категории предусматривается комната
площадью 15-20 м2, а для третьей категории - две комнаты общей
площадью 30-40 м2.
Для котлов
группы III при значительных размерах котельных (суммарная производительность
котлов более 100 т/ч) в отдельных случаях по решению проектных организаций
можно предусмотреть, кроме основной лаборатории, также экспресс-лабораторию -
отдельную комнату площадью 15-20 м2.
2.2.9. Водные
лаборатории желательно располагать в непосредственной близости к общекотельному
водному щиту.
2.2.10. В
водной лаборатории третьей категории предусматривается организация
приточно-вытяжной вентиляции, желательно с кондиционированием воздуха.
2.2.11. В лабораториях всех трех категорий вода подводится из
хозяйственно-питьевого водопровода, устанавливается водопроводная раковина и
предусматривается канализация.
В лабораториях
всех категорий должны быть предусмотрены установка светильников дневного света
и подвод электроэнергии со стабилизированным напряжением. Сеть должна быть
рассчитана на одновременную работу всех электроприборов, указанных в табл. 3.
2.2.12.
Необходимый минимум оборудования и приборов приведен в табл. 3.
В помещение
лабораторий второй и третьей категорий для производства аналитических работ
должен быть подведен трубопровод конденсата пара.
Для отбора
среднесуточных проб химически обработанной и питательной воды трубопроводы
указанных отборов проб должны быть подведены в помещение лабораторий всех трех
категорий. Кроме того, должен быть организован непрерывный отбор этих проб в
бутыли вместимостью 25 л. Бутыли устанавливают в специальном закрытом на замке
боксе.
2.3. Требования к
предприятиям, эксплуатирующим котлы.
2.3.1. Должно
быть организовано проведение анализов в объеме химического контроля и по
методикам, предусмотренным настоящим РТМ, а также организован учет результатов
анализов с выводом среднемесячных показателей, по которым проводится
сопоставление с требованиями по водному режиму, обусловленными соответствующими
нормативными общесоюзными и ведомственными документами.
2.3.2. Для
осуществления представительного непрерывного химического контроля за водным
режимом в котельных всех типов рекомендуется производить отбор среднесуточных
проб химически обработанной и питательной воды, анализы которых наиболее полно
характеризуют надежность водного режима котельных агрегатов.
2.3.3. В объем
химического контроля, предусмотренный настоящим РТМ, входит определение
прозрачности всех вод для косвенного контроля за содержанием взвешенных
веществ. Учитывая простоту и несложность анализа, а также то, что назначение
определения прозрачности - предотвращение заноса трубодисперсными соединениями
поверхностей нагрева котла и что в условиях промышленной энергетики при
отсутствии налаженной предочистки такие заносы возможны, данное определение
рекомендуется выполнять во всех типах котельных.
2.3.4. На
основании конкретных условий работы котельной и указаний настоящего РТМ должен
быть установлен необходимый объем химического контроля, реализована схема
отбора проб и оборудована водная лаборатория. В случае необходимости это
осуществляется с привлечением специализированной организации.
2.3.5. При
монтаже трубопроводов для отбора проб воды и пара должен быть выдержан уклон в
сторону движения пробы. Трубопроводы не должны изолироваться независимо от их
длины, и для обеспечения безопасности должны быть ограждены.
2.3.6. Для
котлов групп II и III должен поддерживаться расчетный расход проб, который для
питательной воды и пара составляет 20-30 кг/ч, а для котловой воды - 30-40
кг/ч. Для котлов группы I расчетный расход поддерживается только в момент
отбора пробы.
2.3.7. Для
котлов группы I все аналитические определения производят с помощью
экспресс-лаборатории типа ЭЛВК-5 и полуавтоматического кислородомера типа ОКВ
производства завода «Лаборприбор» в г. Клин. Все необходимые растворы для этих
определений приготовляют в центральной заводской лаборатории, а в случае ее
отсутствия готовят на месте из фиксаналов.
2.3.8. В
водной лаборатории второй категории для котлов группы II выполняют все анализы
и определения, предусмотренные в табл. 1 для котлов этой группы. При наличии
центральной заводской лаборатории в водной лаборатории этой категории можно
обойтись без нагревания и взвешивания на аналитических весах.
Солесодержание
определяют электрометрически лабораторным солемером (например, с использованием
мегомметра). Тарирование его осуществляет центральная заводская лаборатория или
водная лаборатория специализированной энергетической организации.
Определение
растворенного кислорода осуществляют лабораторным полуавтоматическим
кислородомером типа ОКВ.
Лаборатория
оборудуется в соответствии с п. 2.2.11.
2.3.9. В
соответствии с п. 2.2.8 для котлов группы III предусматривается
водная лаборатория, состоящая из двух комнат. Одна из них служит для тонких
аналитических операций с использованием аналитических весов. В таких
лабораториях необходимо организовать нагрев, сушку и приготовление химически
обессоленной воды с «нулевым» содержанием солей жесткости и соединений железа.
В лаборатории проводят определения содержания фосфатов, аммиака, свободной
углекислоты и соединений железа. В зависимости от особенностей технологической
схемы водоподготовки в лаборатории может потребоваться определение
окисляемости, содержания нитратов, меди, сульфитов и выполнение полного анализа
воды по упрощенной схеме.
В лаборатории
необходимо создать условия для быстрого охлаждения проб и их нагрева.
Все приборы
экспресс-контроля в целях обеспечения бесперебойности их действия
дублируют.
2.3.10. Должны
быть созданы все условия для получения предварительной пробы при ее отборе на
анализ.
Таблица 4
|
Наименование аналитических определений
|
Категория лабораторий
|
|
Первая
|
Вторая
|
Третья
|
|
Прозрачность и взвешенные вещества
|
+
|
+
|
+
|
|
Щелочность
|
+
|
+
|
+
|
|
Жесткость
|
+
|
+
|
+
|
|
Хлориды
|
+
|
+
|
+
|
|
Кислород растворенный
|
+1
|
+
|
+
|
|
Солесодержание
|
-
|
+
|
+
|
|
Фосфаты
|
-
|
-
|
+
|
|
Нитраты
|
-
|
+2
|
+2
|
|
Углекислота свободная
|
-
|
+
|
+
|
|
Соединения железа
|
-
|
+
|
+
|
|
рН
|
-
|
+
|
+
|
|
Аммиак
|
-
|
+2
|
+2
|
1 Только при наличии
обескислороживания питательной воды.
2
Только при наличии аминирования или нитратирования.
При отборе
пробы, анализируемой на содержание соединений, находящихся частично в
грубодисперсной форме (железо), пробоотборную трассу периодически продувают с
максимально возможной интенсивностью. По окончании продувки устанавливают
необходимый расход и температуру 20-40 °С испытуемой воды, затем не ранее чем через 3 ч отбирают пробы. Необходимым условием
представительности отбора в этом случае является непрерывное действие
пробоотборных точек.
При отборе и
транспортировке пробы создаются условия, исключающие возможность загрязнения
пробы из окружающей среды. Пробы питательной воды, конденсата и пара отбираются
в полиэтиленовые сосуды.
Кроме того,
выполняются все условия, предусмотренные в подразделе 13.3.
2.3.11. В
лабораториях трех категорий должна быть организована возможность аналитических
определений в соответствии с табл. 4.
Кроме
определений, перечисленных в табл. 4, в отдельных случаях в лабораториях второй
и третьей категорий при наличии соответствующих загрязнений в питательной воде
производят определение содержания нитритов, веществ, экстрагируемых эфиром
(нефтепродуктов), меди и окисляемости воды. Необходимость этого в каждом
отдельном случае устанавливает специализированная наладочная организация. В
соответствии с этим используются ведомственные методические указания1.
2.3.12. При
организации аналитических определений рекомендуется использовать методы,
перечисленные в табл.
5 и изложенные в последующих разделах.
В табл. 5
также приведены чувствительность указанных методов определения и пределы
округления, которые рекомендуются при расчете результата анализов.
3.1.
Прозрачность столба предварительно тщательно перемешанной пробы воды дает
возможность приближенно оценить содержание в ней взвешенных веществ.
Наиболее
простой метод определения прозрачности - установление момента исчезновения видимости
опускаемого в воду кольца Ø 20 мм, изготовленного из проволоки Ø
2 мм и укрепленного на металлической линейке с сантиметровой шкалой.
3.2.
Проволочное кольцо из черной проволоки заданных размеров с металлической
линейкой опускают в стеклянный цилиндр, заполненный испытуемой водой, до тех
пор, пока контуры кольца сделаются невидимыми.
Таблица 5
|
Наименование аналитических определений
|
Рекомендуемый метод
определения
|
Единица измерения
|
Чувствительность
|
Предел округления
|
|
Прозрачность
|
«По кольцу», «По шрифту»
|
см
|
1
|
1
|
|
Взвешенные вещества
|
Косвенно по прозрачности
|
мг/кг
|
5
|
5
|
|
Щелочность вод типа конденсата
|
Титрование кислотой
|
мг-экв/кг
|
0,05
|
0,05
|
|
Щелочность прочих вод
|
«
|
«
|
0,1
|
До двух значащих цифр
|
|
Жесткость меньше 20 мкг-экв/кг
|
Трилонометрический, колориметрический
|
мкг-экв/кг
|
2
|
1
|
|
Жесткость больше 0,02 мг-экв/кг
|
Трилонометрический титрованием
|
мг-экв/кг
|
0,01
|
До двух значащих цифр
|
|
Хлориды
|
Аргентометрический
|
мг/кг
|
1
|
«
|
|
Кислород растворенный
|
Индигокарминовый
|
мкг/кг
|
10
|
10
|
|
Солесодержание
|
Электрометрический
|
мг/кг
|
0,1
|
До двух значащих цифр
|
|
Фосфаты в котловой воде
|
Молибдатный
|
«
|
1
|
1
|
|
Нитраты в котловой воде
|
Ионитный
|
«
|
10
|
10
|
|
Углекислота свободная
|
Титрование щелочью
|
«
|
1
|
До двух значащих цифр
|
|
Соединения железа
|
Сульфосалициловый
|
мкг/кг
|
50
|
50
|
|
Аммиак
|
Титрованием и по Несслеру
|
мг/кг
|
0,2
|
До двух значащих цифр
|
|
рН
|
По универсальному индикатору
|
-
|
1
|
1
|
Примечание. При необходимости определения содержания нитритов,
соединений меди или эфирорастворимых веществ (нефтепродуктов) для определения
осуществляют по методическим указаниям ведомственных нормативных материалов
Минэнерго.
Глубина
погружения кольца в сантиметрах соответствует числовому значению прозрачности
воды «по кольцу». В табл. 6 приведены значения этого показателя (см),
пересчитанные на прозрачность «по шрифту», фигурирующую в нормах ОСТ 24.034.02.
3.3.
Установить зависимость между прозрачностью и концентрацией взвешенных веществ
можно следующим образом.
Для данной
исходной воды с привлечением специализированной наладочной организации
производят ряд анализов с весовым определением концентрации взвешенных веществ
(по разности сухих остатков фильтрованной и нефильтрованной воды). Одновременно
производят в каждом случае определение прозрачности воды. Все результаты
анализов по параллельным пробам наносят на координатные оси и по полученным
точкам вычерчивают кривую.
Менее точный,
но более быстрый метод - построение кривой по данным одной пробы воды в период
ее максимального загрязнения взвешенными веществами. Получение ряда точек по
прозрачности достигается последовательным разбавлением первоначальной,
тщательно перемешанной пробы дистиллатом с определением прозрачности каждого
нового эталона.
Таблица 6
|
"По кольцу"
|
"По шрифту"
|
"По кольцу"
|
"По шрифту"
|
|
2
|
0,5
|
21
|
15
|
|
3
|
1
|
22
|
16
|
|
4
|
2
|
24
|
17
|
|
6
|
3
|
26
|
18
|
|
7
|
4
|
28
|
19
|
|
8
|
5
|
29
|
20
|
|
10
|
6
|
30
|
21
|
|
11
|
7
|
31
|
22
|
|
12
|
8
|
32
|
23
|
|
13
|
9
|
33
|
24
|
|
15
|
10
|
34
|
25
|
|
16
|
11
|
36
|
26
|
|
17
|
12
|
37
|
27
|
|
19
|
13
|
38
|
28
|
|
20
|
14
|
41
|
30
|
Для
определения прозрачности воды, прошедшей химическую обработку (например, после
осветлителя в процессе известкования), в качестве исходной воды для
приготовления эталонов должна быть взята именно эта вода в период ее низкой
прозрачности.
4.1.
Определение щелочности воды основано на титровании растворенных в воде щелочных
соединений кислотой в присутствии индикаторов, меняющих свою окраску в
зависимости от реакции среды.
При титровании
с метилоранжем или смешанным индикатором определяется общая щелочность, условно
характеризующая суммарное содержание бикарбонатов, карбонатов, гидратов, 2/3
ортофосфатов и гуматов.
При титровании
с фенолфталеином определяются полностью гидраты, 1/2 карбонатов, 1/3
ортофосфатов и гуматы.
Данные по
изменению цвета индикаторов в зависимости от рН среды приведены в табл. 7.
4.2.
Необходимые реактивы:
серная кислота
или соляная - 0,1 н. раствор;
серная кислота
или соляная - 0,01 н. раствор;
фенолфталеин -
1 %-ный спиртовый раствор;
метилоранж -
0,1 %-ный водный раствор;
смешанный
индикатор: смесь равных объемов спиртовых растворов метилового красного - 0,25
% и метиленового голубого - 0,17 %.
4.3. Ход определения.
4.3.1. Для
определения щелочности исходной известкованной, коагулированной,
катионированной и питательной воды 100 мл испытуемой воды помещают в коническую
колбу, прибавляют одну-две капли фенолфталеина и в случае появления розовой
окраски титруют 0,1 н. кислотой до обесцвечивания. Отметив расход кислоты,
добавляют две-три капли метилоранжа или смешанного индикатора и продолжают
титрование до изменения окраски.
Если проба
после добавления фенолфталеина не изменила окраски, сразу прибавляют метилоранж
или смешанный индикатор и титруют 0,1 н. кислотой до перехода окраски.
Количество миллилитров 0,1 н. кислоты, израсходованной на титрование по
фенолфталеину, численно соответствует щелочности воды по фенолфталеину в
миллиграмм-эквивалентах на килограмм. Количество миллилитров 0,1 н. кислоты,
израсходованной суммарно на титрование пробы по фенолфталеину и метилоранжу или
фенолфталеину и смешанному индикатору, численно соответствует общей щелочности
воды в миллиграмм-эквивалентах на килограмм.
Таблица 7
|
Индикаторы
|
Окраска при реакции среды
|
Интервал перехода рН
|
|
кислой
|
щелочной
|
нейтральной
|
|
Фенолфталеин (ф. ф.)
|
Бесцветная
|
Фиолетовая
|
Бесцветная
|
8,2 - 10
|
|
Метилоранж (м. о.)
|
Розовая
|
Желтая
|
Оранжевая
|
3,1 - 4,4
|
|
Смешанный
|
фиолетовая
|
Зеленая
|
Грязно-серая
|
4,2 - 6,2
|
4.3.2.
Для определения щелочности котловой воды 15-50 мл котловой воды помещают в
коническую колбу, разбавляют до 100 мл дистиллатом и определяют общую
щелочность титрованием 0,1 н. кислотой в присутствии смешанного индикатора,
дающего более резкий переход окраски в конце титрования.
Количество
миллилитров 0,1 н. кислоты, израсходованной на титрование 50 мл пробы,
умноженное на два, а при титровании 25 мл пробы, умноженное на четыре,
определяет общую щелочность воды в миллиграмм-эквивалентах на килограмм.
4.3.3. Для
определения щелочности вод типа конденсата (при щелочности ниже 0,2 мг-экв/кг)
по 100 мл испытуемой воды помещают в две одинаковые конические колбы,
прибавляют по две-три капли метилоранжа или смешанного индикатора и первую
титруют 0,01 н. кислотой из микробюретки с оттянутым носиком до первого
изменения окраски по сравнению со второй колбой, служащей «свидетелем».
Сравнение производят на белом фоне при сильном освещении.
Количество
миллилитров 0,01 н. кислоты, израсходованной на титрование пробы, деленное на
10, численно равно щелочности воды в миллиграмм-эквивалентах на килограмм.
5.1. Трилон Б
- кислая двузамещенная натриевая соль этилендиаминотетрауксусной кислоты - при
рН > 9 связывает во внутрикомплексные соединения катионы кальция и магния.
Некоторые
красители (кислотный хром темно-синий, эриохром черный ЭТ-00) дают с катионами
солей жесткости непрочные окрашенные соединения красного цвета. При добавлении
в воду с подобными окрашенными соединениями раствора трилона Б в эквивалентной
точке происходит их полное разрушение с изменением окраски раствора в синий
цвет. В присутствии ионов цинка или меди (неотчетливый переход окраски)
определение жесткости производят с прибавлением раствора сульфида натрия,
связывающего эти катионы в нерастворимые сульфидные соединения.
Влияние ионов
марганца, приводящего к быстрому обесцвечиванию окраски, устраняют прибавлением
к пробе раствора солянокислого гидроксиламина.
5.2.
Необходимые реактивы:
трилон Б -
0,05 н. раствор;
трилон Б -
0,005 н. раствор;
аммиачный
буферный раствор: 20 г хлористого аммония (х. ч.) растворяют в дистиллате,
добавляют 100 мл концентрированного раствора аммиака (х. ч., уд. вес 0,91) и
разбавляют до 1 л дистиллатом;
боратный
буферный раствор: 40 г десятиводной буры (х. ч.) растворяют в 800 мл дистиллата
и отдельно растворяют 10 г едкого натра (х. ч.) в 100 мл дистиллата; смешивая
после охлаждения оба раствора, дополняют объем до 1 л дистиллатом;
кислотный хром
темно-синий: 0,5 г индикатора растворяют в 10 мл аммиачного буферного раствора
и разбавляют до 100 мл этиловым спиртом;
эриохром
черный ЭТ-00: 0,5 г индикатора растворяют в 10 мл аммиачного буферного раствора
и разбавляют до 100 мл этиловым спиртом;
сернистый
натрий - 10 %-ный раствор (хранить в полиэтиленовой посуде не более двух
недель);
солянокислый
гидроксиламин - 2 %-ный раствор;
исходная вода,
разбавленная катионированным дистиллатом до жесткости 100 мкг-экв/кг;
смесь
буферного раствора с индикатором в обычном соотношении с прибавлением исходной
воды с жесткостью 100 мкг-экв/кг в количестве, эквивалентном заданному эталону
жесткости;
катионированный
дистиллат, не содержащий солей жесткости.
5.3. Ход определения.
5.3.1. Для
определения жесткости природной, известкованной, коагулированной воды 100 мл
пробы помещают в коническую колбу, прибавляют 5 мл аммиачного буферного
раствора, семь капель индикатора кислотного хрома темно-синего и медленно титруют
при постоянном перемешивании 0,05 н. раствором трилона Б до отчетливого
изменения окраски в синий цвет.
Количество
миллилитров 0,05 н. трилона Б, пошедшего на титрование пробы,
деленное на два, определяет общую жесткость воды в миллиграмм-эквивалентах на
килограмм.
При нечетком
переходе окраски или ее обесцвечивании определение повторяется с
предварительным прибавлением 0,5 мл раствора сернистого натрия для устранения
мешающего действия ионов меди и цинка или с прибавлением трех капель раствора
солянокислого гидроксиламина для устранения мешающего действия соединений
марганца.
5.3.2. Для вод
с жесткостью ниже 100 мкг-экв/кг 100 мл пробы помещают в коническую колбу,
прибавляют 5 мл аммиачного буферного раствора, семь капель индикатора
кислотного хрома темно-синего и медленно титруют при постоянном перемешивании
0,005 н. раствором трилона Б из микробюретки с размером капли не более 0,05 мл
до изменения окраски.
Количество
миллилитров 0,005 и трилона Б, пошедшего на титрование пробы, умноженное на 50,
определяет общую жесткость воды в микрограмм-эквивалентах на килограмм.
5.3.3. Для вод
с жесткостью ниже 20 мкг-экв/кг (колориметрический вариант) приготавливают два
ряда эталонов воды с жесткостью 0; 1; 5 и 10 мкг-экв/кг путем разбавления 0; 1;
5 и 10 мл эталонной жесткой воды до 100 мл дистиллатом в два ряда конических
колб.
В первый ряд
эталонов прибавляют до 5 мл аммиачного буферного раствора и по семь капель
кислотного хрома темно-синего.
Во второй ряд
эталонов прибавляют по 1 мл боратного буферного раствора и по семь капель
эриохрома черного ЭТ-00.
В результате
сравнения гаммы окрасок в обоих рядах эталонов выбирают буфер и индикатор,
которые для данной исходной воды дают более постепенный и растянутый переход
окраски от синего к розовому цвету. Это является индивидуальной особенностью
данной воды и обусловлено спецификой ее «солевого букета».
Из выбранной
пары (красителе и буфера) готовят рабочий ряд эталонных растворов с
жесткостями: 0; 1; 3; 5; 7; 10; 15 и 20 мкг-экв/кг путем разбавления 0; 1; 3;
5; 7; 10; 15 и 20 мл эталонной жесткой воды до 100 мл дистиллатом.
Одновременно
этот буфер и краситель в тех же количествах прибавляют к 100 мл испытуемой воды
и сравнивают с окраской шкалы эталонов, которую каждый раз готовят вновь.
Окраска пробы,
совпадающая с эталоном, непосредственно определяет величину жесткости в
микрограмм-эквивалентах на килограмм.
5.3.4.
Сравнение окраски испытуемой воды с окраской эталонов позволяет определить
фактическое значение жесткости с чувствительностью 0,5-2 мкг-экв/кг (в
зависимости от специфических особенностей «солевого букета» данной воды).
В случае
необходимости определение ведется с вводом в испытуемую воду растворов
сернистого натрия или солянокислого гидроксиламина для связывания катионов,
мешающих определению.
При анализе малых
жесткостей большую ошибку может дать загрязнение солями жесткости буферных
растворов: аммиачного или боратного. Отсутствие подобного загрязнения или
размер необходимой поправки устанавливают сравнением со шкалой эталонов
интенсивности окраски испытуемой воды с красителем при однократном или
двукратном количестве аммиачного буферного раствора или боратного против
прописи.
При наличии
лабораторных катионитных фильтров можно ликвидировать загрязнение буферных
растворов, пропуская их через эти фильтры, заряженные одноименным ионом.
5.3.5. Для
качественного сравнения фактической жесткости с данным эталоном к 100 мл пробы
добавляют смесь буферного раствора с индикатором и эталоном жесткости в обычном
количестве. Если появляется красная окраска, то жесткость воды выше эталона
(например, 50 мкг-экв/кг для отключения катионитного фильтра), если синяя, то
жесткость воды не достигла заданного предела.
6.1. После
связывания хлор-ионов азотнокислым серебром образуется хлористое серебро (белый
осадок), при этом первые избыточные капли реактива в присутствии хромовокислого
калия в нейтральной (по фенолфталеину) среде приводят к появлению бурой окраски
хромовокислого серебра.
6.2.
Необходимые реактивы:
серебро
азотнокислое - 0,0282 н. раствор (хранить в коричневой склянке);
кислота серная
- 0,1 н. раствор;
калий
хромовокислый - 10 %-ный раствор;
фенолфталеин -
1 %-ный спиртовый раствор.
6.3. Ход определения.
6.3.1.
Отбирают пробу воды в зависимости от предполагаемого содержания хлоридов в
объеме, указанном в табл. 8.
Пробу помещают
в коническую колбу, прибавляют две капли фенолфталеина и нейтрализуют серной
кислотой до исчезновения розовой окраски. Затем добавляют 1 мл раствора
хромовокислого калия и титруют из бюретки азотнокислым серебром до появления
устойчивой бурой окраски. При объеме пробы более 100 мл ее предварительно
упаривают.
Таблица 8
При
объеме пробы 100 мл количество миллилитров азотнокислого серебра после
вычитания поправки на окраску, принимаемой равной 0,2 мл, и умножения на 10
соответствует содержанию хлоридов в миллиграммах на килограмм.
При ином
объеме пробы результат умножают на корректирующий коэффициент К,
приведенный в табл.
8.
6.3.2. При
большом числе определений в лаборатории должен быть сосуд для сбора
оттитрованных проб и регенерации серебра.
6.3.3.
Допускается применение меркуриметрического метода по «Инструкции по анализу
воды, пара и отложений в теплосиловом хозяйстве» при условии соблюдения правил
техники безопасности при работе с ртутными соединениями.
7.1. Сухой
остаток определяют путем выпаривания определенного объема профильтрованной
пробы воды во взвешенной фарфоровой чашке и высушиванием остатка при
температуре 105 °С до постоянного веса.
Для установок
с прямоточной схемой водоподготовки натрийкатионитным методом двухступенчатого
умягчения прямое определение сухого остатка котловой воды может быть заменено
косвенным определением его по общей щелочности котловой воды. Зависимость
солесодержания от щелочности S = f(Щ)
может быть установлена как расчетным путем по анализу исходной воды, так и
путем ряда весовых анализов котловой воды.
Для исходных
вод с содержанием хлоридов более 5 мг/кг определение сухого остатка котловой
воды может достаточно точно производиться косвенно по концентрации хлор-иона.
Зависимость солесодержания от концентрации хлор-иона S = f[С1-]
устанавливается экспериментальным путем на основании нескольких анализов
котловой воды с параллельным определением сухого остатка и содержания хлоридов.
Весовой метод
определения сухого остатка ввиду значительной длительности применяют как
контрольный для тарирования приборов-солемеров, определяющих сухой остаток
косвенным методом по электропроводности раствора.
7.2. Используя
электропроводность ионов, можно приближенно определить концентрацию
ионодисперсных загрязнителей. Эквивалентные электропроводности большинства
ионов имеют достаточно близкое значение. Исключением являются ионы H+ и ОН-. Поэтому
сильнощелочные воды перед электрометрическим определением должны
нейтрализоваться до значения рН 6-8.
Для
электрометрического определения могут использоваться различные солемеры,
выпускаемые промышленностью, например: солемеры, разработанные ЦЛЭМ Тулэнерго,
с интервалом измерения 0,02-2,5 мг/кг, солемеры Ленинаканского завода
аналитических приборов с пределом измерений 0-500 мг/кг.
Наиболее
дешевым, доступным и универсальным является лабораторный солемер конструкции
Центроэнергочермета с датчиком, изготовляемым на месте по эскизу (см. чертеж) с
использованием в качестве измерительного прибора переносного мегомметра типа
М-1101М, М-1102/1 или МС-05.
Расстояние между электродами L в зависимости от
температуры, удобной для работы в данной лаборатории:
Температура, °С 20 25 30 35
L, мм 97 108,3 120,8 132
Мегомметр
предварительно тарируют, т. е. определяют, какому солесодержанию соответствует
показание шкалы мегомметра в мегаомах. Датчик снабжен термометром, электродами
служит проволока из нихрома Ø 0,5 - 1,0 мм. В оба конца стеклянной
трубки вставлены электроды, которые при помощи небольших кусков провода (в
резиновой или пластиковой оплетке) присоединяются к клеммам мегомметра. Концы
электрода загибают в виде кольца. В качестве датчика для питательной воды может
быть использована обычная бюретка на 25 мл с диаметром примерно 12 мм. Для
котловой воды и пара диаметр трубки равен примерно 4 мм. Общая длина трубки -
200-250 мм.
Для
определения солесодержания конденсата пара солемер градуируют расчетным путем,
а для определения питательной и котловой воды - по стандартным растворам воды
данного состава и с сухими остатками, определенными весовым способом.
Периодически
(не реже одного раза в месяц) градуировку солемера проверяют по сухим остаткам,
определенным весовым способом.
7.3.
Градуировку солемера для конденсата пара производят расчетным путем.
Солесодержание пробы в пересчете на NaCl вычисляют по формуле

где SNaCl - солесодержание в пересчете на NaCl,
мг/кг; L - расстояние между электродами, см; R
-
сопротивление по мегомметру, МОм; f - площадь токопроводящего
сечения датчика, см2; kNaCl
- удельная электропроводность раствора, содержащего 1 мг/кг NaCl при
определенной температуре, Мсим/см.
Значения kNaCl при данных температурах:
Температура, °С 15 18 20 25 30 35 40
kNaCl,
Мсим/см 1,75 1,854 1,94 2,16 2,39 2,62 2,86
Для
датчика с расстоянием между электродами L = 9,4 см, соответствующей t
= 18 °С
и с диаметром электрода d = 4 мм, f
= 0,1256 см2
расчетная формула может быть упрощена:
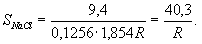
Числитель формулы в зависимости от температуры имеет следующие значения:
Температура, °С 15 18 20 25 30 35 40 45
Значение числителя 43,3 40,3 38,5 35,0 30,9 27,6 24,7 22,1
По
результатам расчетов строят тарировочные кривые для различных температур.
Солесодержание откладывают по оси ординат, а сопротивление в мегаомах - по оси
абсцисс.
Датчик солемера:
1 - резиновые трубки; 2
- шкала подвижного электрода; 3 - выход пробы; 4 - колено; 5,
15 - верхняя и нижняя головки из оргстекла; 6 - ртутный
термометр; 7, 17 - подвижный и неподвижный электроды из
нихромовой проволоки d = 0,5...1,0 мм; 8 -
вход пробы; 9 - воронка; 10 - скоба; 11 - подводящая
стеклянная трубка; 12- панель 100´350 мм; 13 -
стеклянная трубка датчика; 14 - резиновая пробка; 16 - болт М5; 18
- зажим пружинный
Вместо
тарировочных кривых можно пользоваться универсальной таблицей1 (табл. 9). При
пользовании табл.
9 следует учитывать возможность переноса запятой с учетом обратной
пропорциональности S и R, а также необходимость
выполнения требования по выдерживанию расстояния между элементами датчика.
7.4.
Градуировку солемера для котловой воды производят по стандартным растворам,
приготовленным из котловой воды с максимальным солесодержанием.
Котловую воду отбирают из котла, когда
солесодержание ее наиболее высокое (по хлоридам или щелочности). Объем исходной
воды должен быть 5 - 8 л. Для увеличения солесодержания применяют упаривание.
В исходной
пробе котловой воды определяют сухой остаток, щелочность по фенолфталеину и
метилоранжу. Из исходной котловой воды с известным сухим остатком готовят
стандартные растворы путем соответствующего разбавления ее чистым конденсатом.
Стандартные растворы готовят с сухими остатками 500, 1000, 1500, 2000, 2500,
3000 мг/кг в зависимости от желаемого диапазона измерений. Пробы котловой воды
перед заливкой их в датчик должны быть нейтрализованы по фенолфталеину 0,5
%-ным раствором соляной кислоты.
По результатам
градуировки строят график, где по оси ординат откладывают солесодержание, а по
оси абсцисс - показание мегомметра. Сопротивление определяют при одной и той же
температуре стандартных растворов t = 15 °С.
Допускается
использовать ручную компенсацию температуры путем изменения расстояния L по
чертежу.
7.5. Градуировку
солемера для питательной воды производят по стандартным растворам,
приготовленным из исходной химически очищенной воды с известным
солесодержанием, определенным весовым способом.
Стандартные
растворы готовят с солесодержанием 30, 50, 70, 100, 150, 200, 250, 300 мг/кг в
зависимости от необходимого диапазона солесодержания.
Условия
градуировки и техника ее проведения такие же, как и при градуировке солемеров
для котловой воды.
7.6. Указания по работе с
лабораторными солемерами.
7.6.1. Датчик
устанавливают на расстоянии одного метра от мегомметра.
7.6.2.
Присоединение датчика осуществляют проводами в резиновой или пластикатовой
оплетке.
7.6.3. Перед
определением солесодержания котловой воды ее предварительно нейтрализуют 0,5
%-ной соляной кислотой по фенолфталеину.
7.6.4.
Проверяют исправность прибора:
вращают
рукоятку мегомметра при разомкнутых клеммах; стрелка мегомметра устанавливается
на «∞»;
замыкают
клеммы накоротко куском медной проволоки; стрелка при вращении рукоятки
устанавливается на 0 шкалы;
присоединяют
мегомметр к датчику, не заполненному водой, и проверяют на отсутствие
заземления; стрелка при вращении рукоятки устанавливается на «∞».
7.7. Ход определения.
7.7.1. Датчик
предварительно промывают конденсатом и испытуемой водой. Затем наполняют датчик
испытуемой водой, замеряют температуру воды, подключают датчик к мегомметру и
вращают его ручку со скоростью не менее 1,5 - 2 об/сек в течение 2 - 3 сек.
Записывают показания на шкале мегомметра, затем показание прибора по графику
переводят в миллиграммы на килограмм.
7.7.2.
Солесодержание конденсата пара определяют после предварительной дегазации
пробы. Пробу конденсата упаривают в четыре раза в плоскодонной колбе из
химически устойчивого стекла. Колба должна быть предварительно обработана кислотой
и проверена на выщелачивание. После упаривания колбу охлаждают до 25-35 °С,
предварительно закрыв пробкой или фильтровальной бумагой для предохранения от
попадания в нее пыли и газов из воздуха. Разделив солесодержание упаренной
пробы на кратность упаривания, находят условное солесодержание.
Таблица 9
Таблица
солесодержания конденсата пара к солемеру с мегомметром типа М-1101 с
дегазацией проб путем упаривания
|
Показание мегомметра R, МОм
|
Температура
пробы, °С
|
|
18-20
|
21-23
|
24-26
|
27-29
|
30-32
|
33-35
|
36-38
|
39-41
|
42-44
|
45-47
|
48-50
|
51-53
|
54-56
|
57-59
|
60-62
|
63-65
|
66-68
|
69-71
|
72-75
|
76-79
|
80-83
|
84-87
|
88-91
|
92-95
|
96-100
|
|
Солесодержание
конденсата пара S, мг/кг
|
|
2,0
|
2,6
|
2,5
|
2,3
|
2,2
|
2,1
|
2,0
|
1,9
|
1,8
|
1,7
|
1,6
|
1,5
|
1,5
|
1,4
|
1,5
|
1,3
|
1,2
|
1,2
|
1,1
|
1,1
|
1,02
|
0,98
|
0,94
|
0,90
|
0,87
|
0,83
|
|
2,25
|
2,3
|
2,2
|
2,1
|
1,9
|
1,8
|
1,7
|
1,6
|
1,6
|
1,5
|
1,4
|
1,4
|
1,3
|
1,2
|
1,2
|
1,1
|
1,1
|
1,0
|
1,0
|
0,95
|
0,90
|
0,86
|
0,84
|
0,79
|
0,77
|
0,72
|
|
2,5
|
2,1
|
2,0
|
1,9
|
1,7
|
1,6
|
1,6
|
1,5
|
1,4
|
1,3
|
1,3
|
1,2
|
1,1
|
1,1
|
1,1
|
1,0
|
0,97
|
0,93
|
0,89
|
0,85
|
0,81
|
0,77
|
0,75
|
0,71
|
0,69
|
0,66
|
|
2,75
|
1,9
|
1,8
|
1,7
|
1,6
|
1,5
|
1,4
|
1,3
|
1,3
|
1,2
|
1,2
|
1,1
|
1,0
|
1,0
|
0,95
|
0,91
|
0,88
|
0,84
|
0,81
|
0,77
|
0,74
|
0,70
|
0,67
|
0,64
|
0,62
|
0,59
|
|
3,0
|
1,7
|
1,6
|
1,5
|
1,5
|
1,4
|
1,3
|
1,2
|
1,7
|
1,1
|
1,1
|
1,0
|
0,95
|
0,91
|
0,87
|
0,83
|
0,80
|
0,77
|
0,74
|
0,70
|
0,67
|
0,64
|
0,61
|
0,59
|
0,57
|
0,54
|
|
3,25
|
1,6
|
1,5
|
1,4
|
1,3
|
1,3
|
1,2
|
1,1
|
1,1
|
1,0
|
0,96
|
0,92
|
0,87
|
0,84
|
0,80
|
0,77
|
0,74
|
0,71
|
0,68
|
0,62
|
0,62
|
0,59
|
0,56
|
0,54
|
0,52
|
0,50
|
|
3,5
|
1,5
|
1,4
|
1,3
|
1,2
|
1,2
|
1,1
|
1,0
|
1,0
|
0,94
|
0,90
|
0,86
|
0,81
|
0,78
|
0,74
|
0,71
|
0,69
|
0,66
|
0,63
|
0,60
|
0,57
|
0,54
|
0,52
|
0,50
|
0,48
|
0,46
|
|
3,75
|
1,4
|
1,3
|
1,2
|
1,2
|
1,1
|
1,0
|
0,98
|
0,93
|
0,88
|
0,84
|
0,80
|
0,76
|
0,72
|
0,69
|
0,66
|
0,64
|
0,61
|
0,59
|
0,56
|
0,53
|
0,51
|
0,48
|
0,46
|
0,45
|
0,42
|
|
4,0
|
1,3
|
1,2
|
1,2
|
1,1
|
1,0
|
0,97
|
0,91
|
0,87
|
0,82
|
0,78
|
0,75
|
0,71
|
0,68
|
0,65
|
0,62
|
0,60
|
0,57
|
0,55
|
0,55
|
0,50
|
0,47
|
0,45
|
0,43
|
0,42
|
0,39
|
|
4,25
|
1,2
|
1,2
|
1,1
|
1,0
|
0,96
|
0,91
|
0,86
|
0,82
|
0,78
|
0,74
|
0,70
|
0,66
|
0,64
|
0,61
|
0,58
|
0,56
|
0,54
|
0,52
|
0,49
|
0,47
|
0,44
|
0,42
|
0,40
|
0,39
|
0,37
|
|
4,5
|
1,2
|
1,1
|
1,0
|
0,96
|
0,91
|
0,86
|
0,81
|
0,77
|
0,73
|
0,70
|
0,66
|
0,63
|
0,60
|
0,58
|
0,55
|
0,53
|
0,51
|
0,49
|
0,46
|
0,44
|
0,42
|
0,40
|
0,38
|
0,36
|
0,35
|
|
4,75
|
1,1
|
1,0
|
0,97
|
0,91
|
0,86
|
0,81
|
0,77
|
0,73
|
0,69
|
0,66
|
0,63
|
0,59
|
0,57
|
0,54
|
0,52
|
0,50
|
0,48
|
0,46
|
0,43
|
0,41
|
0,39
|
0,38
|
0,36
|
0,34
|
0,32
|
|
5,0
|
1,0
|
0,98
|
0,97
|
0,87
|
0,82
|
0,77
|
0,73
|
0,69
|
0,66
|
0,62
|
0,59
|
0,56
|
0,54
|
0,51
|
0,49
|
0,47
|
0,45
|
0,43
|
0,41
|
0,39
|
0,37
|
0,36
|
0,34
|
0,32
|
0,30
|
|
5,5
|
0,95
|
0,89
|
0,84
|
0,79
|
0,74
|
0,70
|
0,66
|
0,63
|
0,60
|
0,57
|
0,54
|
0,51
|
0,49
|
0,47
|
0,45
|
0,43
|
0,41
|
0,39
|
0,37
|
0,35
|
0,34
|
0,32
|
0,30
|
0,29
|
0,28
|
|
6,0
|
0,87
|
0,82
|
0,77
|
0,72
|
0,68
|
0,64
|
0,61
|
0,58
|
0,55
|
0,52
|
0,49
|
0,47
|
0,45
|
0,43
|
0,41
|
0,39
|
0,37
|
0,36
|
0,34
|
0,32
|
0,30
|
0,29
|
0,28
|
0,26
|
0,25
|
|
6,5
|
0,80
|
0,75
|
0,71
|
0,66
|
0,63
|
0,59
|
0,56
|
0,54
|
0,50
|
0,48
|
0,45
|
0,43
|
0,42
|
0,39
|
0,38
|
0,36
|
0,34
|
0,33
|
0,31
|
0,29
|
0,28
|
0,26
|
0,25
|
0,24
|
0,23
|
|
7,0
|
0,75
|
0,70
|
0,66
|
0,62
|
0,58
|
0,55
|
0,52
|
0,49
|
0,47
|
0,44
|
0,42
|
0,40
|
0,38
|
0,36
|
0,35
|
0,34
|
0,32
|
0,30
|
0,79
|
0,27
|
0,26
|
0,24
|
0,23
|
0,22
|
0,21
|
|
7,5
|
0,70
|
0,65
|
0,61
|
0,58
|
0,54
|
0,51
|
0,48
|
0,46
|
0,43
|
0,42
|
0,39
|
0,37
|
0,35
|
0,34
|
0,32
|
0,31
|
0,79
|
0,28
|
0,26
|
0,25
|
0,24
|
0,23
|
0,21
|
0,20
|
0,19
|
|
8,0
|
0,65
|
0,61
|
0,58
|
0,54
|
0,51
|
0,48
|
0,45
|
0,43
|
0,41
|
0,39
|
0,35
|
0,34
|
0,33
|
0,31
|
0,30
|
0,29
|
0,27
|
0,26
|
0,25
|
0,23
|
0,22
|
0,21
|
0,20
|
0,19
|
0,18
|
|
8,5
|
0,61
|
0,58
|
0,54
|
0,51
|
0,48
|
0,45
|
0,43
|
0,40
|
0,38
|
0,36
|
0,34
|
0,32
|
0,31
|
0,30
|
0,28
|
0,27
|
0,26
|
0,24
|
0,23
|
0,22
|
0,21
|
0,19
|
0,18
|
0,17
|
0,16
|
|
9,0
|
0,58
|
0,54
|
0,51
|
0,48
|
0,45
|
0,42
|
0,40
|
0,38
|
0,36
|
0,34
|
0,32
|
0,30
|
0,29
|
0,28
|
0,26
|
0,25
|
0,24
|
0,23
|
0,22
|
0,20
|
0,19
|
0,18
|
0,17
|
0,16
|
0,15
|
|
9,5
|
0,55
|
0,51
|
0,48
|
0,45
|
0,43
|
0,40
|
0,38
|
0,36
|
0,34
|
0,32
|
0,30
|
0,29
|
0,27
|
0,26
|
0,25
|
0,24
|
0,23
|
0,22
|
0,20
|
0,19
|
0,18
|
0,17
|
0,16
|
0,15
|
0,14
|
|
10
|
0,52
|
0,49
|
0,46
|
0,43
|
0,40
|
0,38
|
0,36
|
0,34
|
0,32
|
0,31
|
0,29
|
0,27
|
0,26
|
0,24
|
0,23
|
0,77
|
0,21
|
0,20
|
0,19
|
0,18
|
0,17
|
0,16
|
0,15
|
0,14
|
0,13
|
|
11
|
0,47
|
0,44
|
0,42
|
0,39
|
0,37
|
0,35
|
0,33
|
0,31
|
0,29
|
0,28
|
0,26
|
0,25
|
0,23
|
0,22
|
0,21
|
0,20
|
0,19
|
0,18
|
0,17
|
0,16
|
0,15
|
0,14
|
0,13
|
0,13
|
0,12
|
|
12
|
0,43
|
0,40
|
0,38
|
0,36
|
0,34
|
0,32
|
0,30
|
0,28
|
0,27
|
0,26
|
0,24
|
0,23
|
0,21
|
0,20
|
0,19
|
0,18
|
0,17
|
0,17
|
0,15
|
0,15
|
0,14
|
0,13
|
0,12
|
0,11
|
0,10
|
|
13
|
0,40
|
0,37
|
0,35
|
0,33
|
0,31
|
0,29
|
0,27
|
0,26
|
0,25
|
0,24
|
0,22
|
0,20
|
0,19
|
0,19
|
0,18
|
0,17
|
0,16
|
0,15
|
0,14
|
0,13
|
0,12
|
0,11
|
0,11
|
0,10
|
0,09
|
|
14
|
0,37
|
0,35
|
0,32
|
0,30
|
0,28
|
0,27
|
0,25
|
0,24
|
0,23
|
0,21
|
0,20
|
0,19
|
0,18
|
0,17
|
0,16
|
0,15
|
0,15
|
0,14
|
0,13
|
0,12
|
0,11
|
0,10
|
0,10
|
0,09
|
0,08
|
|
15
|
0,34
|
0,32
|
0,30
|
0,28
|
0,27
|
0,25
|
0,24
|
0,22
|
0,21
|
0,20
|
0,19
|
0,18
|
0,17
|
0,16
|
0,14
|
0,14
|
0,13
|
0,13
|
0,12
|
0,11
|
0,10
|
0,10
|
0,09
|
0,08
|
0,07
|
|
16
|
0,32
|
0,30
|
0,28
|
0,27
|
0,25
|
0,24
|
0,22
|
0,71
|
0,20
|
0,18
|
0,17
|
0,16
|
0,15
|
0,14
|
0,14
|
0,13
|
0,12
|
0,12
|
0,11
|
0,10
|
0,09
|
0,09
|
0,08
|
0,07
|
0,07
|
|
17
|
0,30
|
0,28
|
0,27
|
0,25
|
0,23
|
0,22
|
0,21
|
0,20
|
0,19
|
0,17
|
0,16
|
0,15
|
0,15
|
0,14
|
0,13
|
0,12
|
0,12
|
0,11
|
0,10
|
0,09
|
0,09
|
0,08
|
0,07
|
0,07
|
0,06
|
|
18
|
0,29
|
0,27
|
0,25
|
0,23
|
0,22
|
0,21
|
0,20
|
0,19
|
0,18
|
0,16
|
0,15
|
0,15
|
0,14
|
0,13
|
0,17
|
0,11
|
0,11
|
0,10
|
0,09
|
0,09
|
0,08
|
0,07
|
0,07
|
0,06
|
0,05
|
|
19
|
0,27
|
0,25
|
0,24
|
0,22
|
0,21
|
0,20
|
0,19
|
0,18
|
0,17
|
0,15
|
0,14
|
0,14
|
0,13
|
0,12
|
0,11
|
0,11
|
0,10
|
0,09
|
0,09
|
0,08
|
0,08
|
0,07
|
0,06
|
0,06
|
0,05
|
|
20
|
0,26
|
0,24
|
0,23
|
0,21
|
0,20
|
0,19
|
0,18
|
0,17
|
0,16
|
0,15
|
0,14
|
0,13
|
0,12
|
0,11
|
0,11
|
0,10
|
0,09
|
0,09
|
0,08
|
0,08
|
0,07
|
0,06
|
0,06
|
0,05
|
0,04
|
Примечание. Расстояние между электродами в датчике L
(см) должно быть установлено по соотношению L = 10kf,
где k - принятая кратность упаривания; f - площадь сечения датчика,
см2. Кратность упаривания может быть в пределах от 3 до 5,
оптимально - 4.
7.7.3.
На точность определения оказывает влияние температура: с повышением температуры
на 1° электропроводность раствора повышается примерно на 2 %. Измерения
производят при постоянной температуре, соответствующей температуре в процессе
градуировки прибора (обычно 25-30 °С). При необходимости пробу следует
предварительно охлаждать до указанной температуры.
При
использовании датчиков с температурной компенсацией пробы не охлаждают.
8.1.
Восстановленная форма индигокармина - лейкосоединение, имеющее золотисто-желтый
цвет, при окислении за счет растворенного в воде кислорода постепенно меняет
свою окраску до темно-синей.
Количественное
определение по методу, предложенному Р. Л. Бабкиным, основано на визуальном колориметрировании
путем сравнения исследуемой воды с лейкоформой индигокармина и
эталонов-имитаторов переходных тонов окраски. Метод используют для определения
содержания кислорода и пределах 10 - 100 мкг/кг О2.
8.2.
Необходимые реактивы и аппаратура:
раствор
индигокармина с кислотностью 0,1-0,2 н. по фенолфталеину и с титром по
кислороду, примерно равным 0,02 мг/мл О2 (определяется
перманганатометрически);
аммиачный
раствор индигокармина - 0,2 н.;
амальгамированный
цинк в редукторе (бюретка на 25 мл с оттянутым концом в виде капилляра).
Гранулированный цинк промывают 5 %-ным раствором азотной кислоты, затем 10
%-ным раствором азотнокислой ртути (окисной или закисной) до образования на
гранулах слоя блестящей амальгамы, переносят в редуктор, промывают в нем водой
и заполняют аммиачным раствором индигокармина;
раствор
пикриновой кислоты - 0,37 г/кг;
шкала
имитаторов; готовится путем смешивания кислого раствора индигокармина и
пикриновой кислоты в объемах, указанных в табл. 10.
Растворы отмеривают в мерную колбу емкостью 250 мл,
которую затем заполняют до метки дистиллатом.
Шкала эталонов
сохраняет устойчивую окраску в течение недели и хранится в парфюмерных склянках
емкостью около 225 мл. Такая же склянка емкостью 225 мл, используемая для
отбора пробы, должна иметь уплотняющую резиновую прокладку, гарантирующую
герметичность. Склянку ставят в специальное ведерко диаметром 120 мм и высотой
200 мм, чтобы иметь возможность вводить в нее реактивы под слоем воды.
8.3. Ход определения.
8.3.1. Пробу
из пробоотборной точки, охлажденную до температуры не выше 50 °С, через
резиновый шланг со стеклянным наконечником Ø 3 мм направляют в склянку,
помещенную в ведерко. Ожидают, пока вода полностью заполнит склянку и ведерко
на 4 - 5 см выше горлышка склянки. Вынув из склянки наконечник, вводят из
редуктора в нее 1,5 мл аммиачного раствора индигокармина. Закрывают под водой
склянку колпачком с уплотняющей резиновой прокладкой и перемешивают содержимое.
Окраска пробы,
совпадающая со шкалой эталонов, непосредственно даст результат анализа.
8.3.2. Для
выполнения данного анализа целесообразно использовать лабораторный
полуавтоматический кислородомер типа ОКВ. Длительность анализа при
использовании этого переносного прибора не превышает 3 мин.
Таблица 10
|
Концентрация О2,
мкг/кг
|
Объем раствора
индигокармина,
мл
|
Объем раствора пикриновой
кислоты, мл
|
|
0,00
|
0,00
|
1,50
|
|
10
|
0,11
|
1,39
|
|
20
|
0,22
|
1,28
|
|
40
|
0,45
|
1,05
|
|
60
|
0,68
|
0,82
|
|
80
|
0,90
|
0,60
|
|
100
|
1,12
|
0,38
|
9.1.
Определение основано на титровании пробы, не содержащей фосфатов, раствором
однозамещенного фосфата калия в присутствии молибдата аммония и хлористого
олова при кислотности среды (0,5-1,0 н.) до совпадения цвета образующегося в
ней синего комплекса с цветом комплекса испытуемой пробы.
9.2.
Необходимые реактивы:
раствор
молибдата аммония в серной кислоте: 100 мл 10 %-ного раствора молибдата аммония
смешивают с 300 мл 50 %-ной (по объему) серной кислоты;
хлористое
олово - 1 %-ный раствор;
хлористый
натрий - 30 %-ный раствор;
катионированная
вода, упаренная примерно до солесодержания котловой воды;
стандартный
раствор однозамещенного фосфата калия, содержащего 100 мг/кг РО43-.
9.3. Ход определения.
9.3.1. В
коническую колбу (а) емкостью 250 мл помещают 1 - 5 мл испытуемой воды, 1 мл
раствора хлористого натрия и 100 мл дистиллата.
В другую
коническую колбу (б) помещают 1 - 5 мл катионированной воды, 1 мл раствора
хлористого натрия и 100 мл дистиллата.
Обе колбы
подогревают до 50 °С, прибавляют в каждую колбу по 5 мл раствора молибдата
аммония и по 1 мл раствора хлористого олова.
Через 1 мин
пробу (б) начинают медленно, по каплям, титровать стандартным раствором до
достижения одинаковой окраски образующегося синего комплекса с окраской комплекса
в испытуемой пробе (а).
9.3.2.
Количество миллилитров стандартного раствора, израсходованного на титрование,
умноженное на 100 и деленное на объем испытуемой воды, соответствует
концентрации РО43- в миллиграммах на килограмм.
10.1. При
пропускании через лабораторный Н-катионитный фильтр котловой воды фильтрат
имеет кислотность К1 соответствующую в эквивалентах
суммарному содержанию сульфатов, хлоридов и нитратов. Зная аналогичную
кислотность К2 для химически обработанной воды, в
которую ввод нитратов не производится, и установив фактическую степень
упаривания воды в котле по щелочности, определяют концентрацию нитратов по
разности.
10.2.
Необходимые реактивы и аппаратура:
лабораторный
Н-катионитный фильтр, изготовленный из бюретки с объемом катионита 25 мл,
отрегенерированный 5 %-ной соляной кислотой и отмытый от избытка кислоты по
смешанному индикатору до кислотности не выше 0,1 мг-экв/кг;
едкий натр -
0,1 н. раствор;
смешанный
индикатор;
дистиллят или
конденсат.
10.3. Ход определения.
10.3.1.
Определяют щелочность котловой воды Щк.в. и щелочность
химически очищенной воды Щх.в. в средней пробе по смешанному
индикатору.
Рассчитывают
кратность упаривания воды 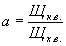 (кратность упаривания
может быть определена и по хлоридам). (кратность упаривания
может быть определена и по хлоридам).
Испытуемую
котловую воду разбавляют в а раз дистиллатом с нейтральной реакцией по
смешанному индикатору.
Пропускают
через Н-катионитный фильтр 200 мл разбавленной котловой воды, спуская в дренаж
первые 50 мл, отбирают 100 мл фильтрата и определяют кислотность К1
по смешанному индикатору титрованием 0,1 н. раствором едкого натра из
микробюретки с размером капли не более 0,05 мл.
Аналогично
определяют кислотность К2 химически обработанной
неразбавленной воды, причем кислотность, соответствующую содержанию сульфатов и
хлоридов.
10.3.2.
Содержание нитратов в котловой воде, выраженное в миллиграммах на килограмм,
определяют по формуле
NO3- = (К1 -
К2) a . 62.
где К1 и К2
- количество миллилитров 0,1 н. раствора едкого натра, пошедшего на
титрование проб; а - кратность упаривания котловой воды; 62 -
эквивалентный вес NO3-.
11.1.
Свободная углекислота количественно связывается децинормальным раствором едкого
натра до бикарбоната натрия; конец реакции фиксируется по индикатору
фенолфталеина.
При
определении СО2 в газоконденсатной смеси из охладителей выпара
деаэраторов или вентиляционных патрубков пароиспользующих теплообменных
аппаратов применяется метод обратного титрования раствора едкого натра, через
который предварительно барботируется конденсатногазовая смесь, подлежащая
анализу.
11.2.
Необходимые реактивы:
едкий натр -
0,1 н. раствор;
фенолфталеин-
1 %-ный спиртовой раствор;
кислота серная
- 0,1 н. раствор.
11.3. Ход определения.
11.3.1. Для
определения СО2 в водах типа конденсата коническую колбу на 500 мл с
нанесенной меткой на уровне 500 мл через резиновый шланг со стеклянным
наконечником полностью заполняют водой из пробоотборной точки с температурой не
выше 50 °С. Колбу тут же закрывают резиновой кососрезанной пробкой и переносят
в лабораторию. Здесь лишнюю воду (сверх отметки 500 мл) осторожно и быстро
сливают и после прибавления 1 мл фенолфталеина осторожно титруют из бюретки 0,1
н. раствором едкого натра, плавно вращая колбу до появления первых следов
розовой окраски, заметных на белом фоне.
Количество
израсходованных миллилитров 0,1 н. раствора едкого натра, умноженное на 8,8,
соответствует содержанию СО2 в миллиграммах на килограмм.
11.3.2. Для
определения СО2 в газоконденсатной смеси в мерный цилиндр на 250 мл
вводят 200 мл 0,1 н. раствора едкого натра. Охлажденную до температуры не выше
40 °С газоконденсатную смесь через резиновый шланг со стеклянным
наконечником из трубки диаметром 3 мм вводят в цилиндр у его днища.
Барботирование газопаровой смеси производится через раствор со скоростью не
выше одного пузырька в секунду и ведется до тех пор, пока объем конденсата и
раствора едкого натра в цилиндре не достигнет 250 мл.
125 мл
полученной смеси титруют из бюретки 0,1 н. раствором серной кислоты в
присутствии фенолфталеина до исчезновения розовой окраски.
11.3.3. Расчет
результата анализа, выраженный в миллиграммах на килограмм СО2,
производят по формуле:
СО2
= (100 - а) 176,
где а - количество
миллилитров 0,1 н. раствора серной кислоты, пошедшего на обратное титрование
125 мл смеси, мл; 176 - коэффициент, учитывающий объем пробы и эквивалент СО2.
12.1. Определение аммиака в
конденсате насыщенного пара.
12.1.1.
Аммиак, присутствующий в виде свободного основания или своей карбонатной соли,
титруют кислотой в присутствии смешанного индикатора.
12.1.2.
Необходимые реактивы:
кислота серная
или соляная - 0,01 н. раствор;
смешанный
индикатор.
12.1.3. Для
определения аммиака 100 мл конденсата помещают в коническую колбу, добавляют
пять капель смешанного индикатора и титруют 0,01 н. кислотой до изменения
окраски от зеленой к фиолетовой.
Количество
миллилитров 0,01 н. кислоты, израсходованной на титрование, умноженное на 1,7,
соответствует содержанию аммиака в миллиграммах на килограмм.
12.2. Определение аммиака в
питательной воде.
12.2.1.
Определение основано на удалении свободного и связанного аммиака четырехкратным
упариванием пробы. Разность щелочности воды без упаривания и после упаривания
соответствует содержанию аммиака.
12.2.2.
Необходимые реактивы:
кислота серная
или соляная - 0,1 н. раствор;
смешанный
индикатор.
12.2.3. Для
определения аммиака в две конические колбы наливают по 100 мл испытуемой воды.
В первой определяют щелочность без упаривания, добавляют пять капель смешанного
индикатора и титруют 0,1 н. раствором кислоты до изменения окраски от зеленой к
фиолетовой; отмечают количество миллилитров кислоты, пошедшей на титрование.
Вторую колбу
ставят на плитку и упаривают примерно в четыре раза, охлаждают и также
определяют щелочность, отмечая расход кислоты.
Разность
расхода 0,1 н. кислоты в миллилитрах при титровании пробы без упаривания и
после упаривания, умноженная на 17, соответствует содержанию аммиака в
миллиграммах на килограмм.
12.3. Контрольный метод с
реактивом Несслера (колориметрический).
12.3.1.
Аммонийные соли со щелочным раствором йодистой ртути (реактивом Несслера) дают
соединение йодистый меркураммоний, окрашенное в желтый цвет.
12.3.2.
Необходимые реактивы:
щелочной
раствор йодистой ртути (реактив Несслера);
сегнетова соль
- 10 %-ный раствор;
Н-катионированная
вода или конденсат, свободный от аммиака;
стандартный
раствор хлористого аммония, содержащий 10 мг/кг NH3-.
12.3.3. Для
определения аммиака в одинаковые пробирки из бесцветного стекла вводят разные
количества стандартного раствора: 0,5; 1,0; 1,5; 2,0 и 2,5 мл, что
соответствует 0,05; 0,01; 0,015; 0,020; 0,025 мг аммиака, доливают до 10 мл
Н-катионированной водой.
В свободную
пробирку наливают 10 мл анализируемой воды. Затем во все пробирки вводят по 0,5
мл раствора сегнетовой соли и по 0,2 мл реактива Несслера. Содержимое пробирок
перемешивают и через 5 мин сравнивают их окраски.
Содержание
аммиака (в миллиграммах на килограмм) в анализируемой пробе определяют
умножением на 100 содержания аммиака в той пробирке, окраска которой совпала с
окраской пробы.
13.1.
Определение основано на образовании в слабощелочной среде окрашенных в
желто-оранжевый цвет комплексов железа с сульфосалициловой кислотой.
Для перевода
всех соединений железа в растворимое состояние пробу вне зависимости от их
предполагаемого содержания предварительно подкисляют и упаривают примерно в 10
раз.
13.2.
Необходимые реактивы:
сульфосалициловая
кислота (30 %-ный раствор) или сульфосалициловый натрий (насыщенный раствор);
серная кислота
- 25 %-ный раствор, не содержащий железа;
персульфат
натрия - 10 %-ный раствор, не содержащий железа;
аммиака - 10
%-ный раствор;
обессоленная
вода или конденсат, не содержащий соединений железа;
стандартный
раствор соли трехвалентного железа, содержащий 10 мг/кг Fe3+.
13.3. Ход
определения.
13.3.1. При
отборе пробы воды соблюдают, по возможности, условия ее представительности по
железу, находящемуся в грубодисперсной форме (короткая трасса, линия отбора из
нержавеющей стали, длительная продувка, достаточность турбулизации потока в
месте отбора, невозможность задержания частиц железа на арматуре или «мертвых»
пространствах пробоподводящей трассы).
Отбор пробы производят
из вертикального нисходящего участка трубопровода. Пробу отбирают в колбу,
предварительно обработанную длительным кипячением с концентрированной соляной
кислотой.
13.3.2. До
отбора пробы в колбу вводят 2 мл 25 %-ной серной кислоты на каждые 100 мл
пробы.
В лаборатории
отмеривают 50 мл пробы, переносят в химический стакан, также обработанный
соляной кислотой, вводят 1 мл персульфата натрия и упаривают примерно в 10 раз.
После этого объем пробы доводят до 25 мл конденсатом (упаривание в два раза).
Одновременно
готовят шкалу эталонных растворов. Для этого стандартный раствор с
концентрацией 10 мг/кг железо разбавляют в колбах в 100, 50, 25, 20, 10 и 5
раз, получают эталонные растворы с концентрацией соответственно 100, 200, 400,
500, 1000 и 2000 мкг/кг железа. Затем в шесть одинаковых пробирок
последовательно вводят по 10 мл эталонных растворов, а в седьмую пробирку - 10
мл подготовленной двукратно упаренной пробы воды.
В каждую
пробирку вводят по 1 мл сульфосалициловой кислоты и по 2 мл раствора аммиака.
После перемешивания содержимого пробирок через 10 мин сравнивают интенсивность
окраски испытуемой воды с окраской эталонных растворов, рассматривая растворы
сверху вниз на белом фоне.
13.3.3.
Содержание железа (в миллиграммах на килограмм) в исходной воде вычисляют путем
деления на два концентрации железа в пробирке эталонной шкалы, окраска которой
соответствует окраске испытуемой двукратно упаренной пробы.
14.1.
Некоторые органические вещества-красители меняют свою окраску в определенном
диапазоне изменений рН.
Используемые
для определения щелочности растворы индикаторов (метилоранж, смешанный
индикатор, фенолфталеин) изменяют свою окраску в диапазонах рН, указанных в табл. 11.
Универсальная
индикаторная бумага, пропитанная растворами различных индикаторов, меняет свою
окраску через переход ряда цветов, воспроизведенных в специальной эталонной
шкале.
14.2.
Необходимые реактивы:
универсальная
индикаторная бумага (рН 1 - 10);
растворы
индикаторов: фенолфталеина, метилоранжа и смешанного индикатора, используемых
для определения щелочности (разд. 4).
14.3. Для
определения значения рН в испытуемую воду опускают универсальную индикаторную
бумагу. Ее окраску сравнивают со шкалой эталонов, имеющейся в пакете для этой
бумаги.
Правильность
показаний индикаторной бумаги можно проверить путем контрольного определения рН
с применением растворов индикаторов, используя при этом данные табл. 11.
Таблица 11
|
Индикатор
|
Окраска индикатора при рН
|
|
Меньше 5
|
5-6
|
6-7
|
7-8
|
8,5
|
Больше 9,0
|
|
Фенолфталеин
|
Бесцветная
|
Бесцветная
|
Бесцветная
|
Бесцветная
|
Слабо-розовая
|
Ярко-розовая
|
|
Смешанный
|
Фиолетовая
|
Грязно-серая
|
Слабо-зеленая
|
Ярко-зеленая
|
Ярко-зеленая
|
Ярко-зеленая
|
|
Метилоранж
|
Розовая
|
Оранжевая
|
Соломенная
|
Желтая
|
Желтая
|
Желтая
|
15.1.
Приготовление обессоленной воды лучше всего осуществлять из конденсата пара в
ионитовом фильтре смешанного действия. Подобный фильтр можно приготовить из
обычной бюретки на 100 мл.
Функции
дренажной системы фильтра и предвключенного механического фильтра выполняют
тампоны гигроскопической ваты.
15.2. В
качестве катионита используют КУ-2, а в качестве анионита - АВ-17 в отношении 1
: 2. Первый предварительно регенерируют в подобном же фильтре 5 %-ной соляной
кислотой, а второй - 2 %-ным раствором едкого натра. Иониты, отмытые
конденсатом от избытка кислоты и щелочи, выгружают в колбу с конденсатом,
перемешивают в ней и совместно загружают в ионитовый фильтр смешанного
действия. Отмывка последнего ведется конденсатом пара.
Оптимальное
суммарное количество ионитов - 100 мл. Получение обессоленной воды ведется со
скоростью фильтрования 10 - 20 м/ч.
15.3. Электропроводность
обессоленного конденсата должна составлять примерно 0,14 . 10-6
сим . см-1.
Для защиты
полученного конденсата от контакта с атмосферой необходимо соблюдать меры
предосторожности. Хранить в полностью заполненной полиэтиленовой посуде с
закрытой пробкой.
16.1.
Приготовление стандартных и титрованных растворов производят по данным табл. 12.
Концентрация
титрованных растворов должна точно соответствовать заданной нормальности.
Для
приготовления титрованных растворов рекомендуется брать навеску или количество
исходного вещества в миллилитрах на 0,5 - 1 % больше, чем по расчету.
Установку
титра производят по результатам не менее чем трех параллельных определений;
берут среднее арифметическое из сходящихся с точностью до 0,1 - 0,2 %
результатов.
16.2.
Нормальность определяемого раствора рассчитывают по формуле
 , ,
где Н1 -
нормальность известного раствора; V1 - количество раствора, нормальность
которого известна, мл; V2 - количество раствора,
нормальность которого определяют, мл.
После
установки нормальности такой раствор легко разбавить дистиллатом до требуемой
концентрации. Количество дистиллата X, добавляемого на каждый литр
раствора, рассчитывают по формуле:
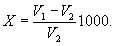
Таблица 12
|
Рабочий раствор
|
Концент-
рация
|
Исходное вещество
|
Обработка исходного
вещества
|
Количество исходного
вещества,
г (мл)
|
Процесс приготовления
|
Проверка титра
|
|
исходное вещество
|
процесс установки титра
|
|
Серная кислота
|
0,1 н.
|
Фиксанал
|
|
1 ампула
|
Перенести содержимое одной ампулы в мерную литровую колбу и разбавить
дистиллатом до метки
|
Не проверяется
|
|
H2SO4,
х. ч., уд. вес 1,84
|
|
2,8 мл
|
В мерную литровую колбу, заполненную до половины дистиллатом, влить
2,8 мл H2SO4, и раствор разбавить дистиллатом до метки
|
0,1 н. раствор углекислого натрия - соды (фиксанал) или 0,1 н. раствор
борнокислого натрия - буры (фиксанал)
|
20 мл 0,1 н. раствора соды (или буры) разбавить до 100 мл дистиллатом
и титровать кислотой в присутствии метилоранжа до оранжевой окраски раствора
|
|
0,01 н.
|
0,1 н. раствор H2SO4
|
|
|
Разбавить 0,1 н. раствор H2SO4 в 10 раз
|
|
|
|
Соляная кислота
|
0,1 н.
|
Фиксанал
|
|
1 ампула
|
|
|
|
|
HCl, х. ч., уд. вес 1,19
|
|
9,0 мл
|
В мерную литровую колбу влить 9 мл HCl и раствор разбавить дистиллатом
до метки
|
То же
|
То же
|
|
0,01 н.
|
0,1 н. раствор HCl
|
|
|
Разбавить 0,1 н. раствор HCl в 10 раз
|
|
|
|
Едкий натр
|
0,1 н.
|
Фиксанал
|
|
1 ампула
|
|
|
|
|
NaOH, х.ч.
|
|
4,5 г
|
Навеску растворить в небольшом количестве дистиллата; раствор
перенести в мерную литровую колбу и разбавить дистиллатом до метки
|
0,1 н. раствор кислоты
|
20 мгл раствора едкого натра, разбавленного дистиллатом до 100 мл,
титровать с метилоранжем или со смешанным индикатором 0,1 н. раствором
кислоты до изменения окраски раствора
|
|
Трилон
|
0,05 н.
|
Фиксанал 0,1 н.
|
|
1 ампула
|
Приготовленный 0,1 н. раствор разбавить дистиллатом в два раза
|
|
|
|
Трилон Б
|
|
10 г
|
Навеску растворить в стакане вместимостью 500 - 800 мл в дистиллате,
отфильтровать, перенести раствор в мерную литровую колбу и разбавить
дистиллатом до метки
|
а) 0,1 н. раствор MgSO4, (фиксанал) или 0,1 н. раствор ZnSO4 (фиксанал). Приготовленный 0,1 н. раствор разбавить дистиллатом в два
раза (0,05 н. раствор)
|
20 мл 0,05 н. раствора сульфата магния или цинка разбавить до 100 мл
дистиллатом, добавить 5 мл аммиачного буферного раствора и семь капель
индикатора хрома темно-синего и титровать трилоном Б до перехода окраски в
синий цвет
|
|
б) Навеску 6,1625 г MgSO4 . 7Н2O (или 7,189 г ZnSO . 7Н2O) растворить в мерной литровой колбе дистиллатом и довести до метки
(0,05 н. раствор)
|
То же
|
|
0,005 н.
|
0,005 н. раствор трилона Б
|
|
|
Разбавить 0,05 н. раствор трилона Б в 10 раз
|
|
|
|
Серебро азотнокислое
|
0,028 н.
|
AgNO3, х.ч.
|
|
4,8 г
|
Навеску растворить в небольшом количестве дистиллата; раствор
перенести в мерную литровую колбу и разбавить дистиллатом до метки
|
NaCl, х. ч., высушенный при температуре 120-150°. Навеску 1,6486 г
растворить в мерной литровой колбе дистиллатом и довести до метки (0,0282 н.
раствор); приготовленный раствор содержит в 1 мл 1 мг Cl-
|
10 мл 0,0282 н. раствора NaCl разбавить до 100 мл
дистиллатом, титровать в присутствии пяти капель хромата калия раствором
азотнокислого серебра до установления устойчивого бурого оттенка
|
|
Индигокармин кислый
|
Титр по кислороду 0,02 мг/мл; кислотность 0,1-0,2 н.
|
Индигокармин
|
Измельчить в ступке
|
0,5 г
|
Навеску нагреть на водяной бане в фарфоровой чашке с 4 мл
концентрированной H2SO4, до полного растворения,
осторожно разбавить дистиллатом до литра. Выдержать пять-шесть дней,
отфильтровать
|
|
|
|
а) Установить титр по кислороду
|
а) 0,1 н. раствор KMnO4 (фиксанал). Приготовленный 0,1 н. раствор разбавить в 10 раз (0,01 н.
раствор KMnO4)
|
а) К 20 мл кислого раствора индигокармина прибавить 50 мл дистиллата
10 мл H2SO4 (1 : 3) и титровать 0,01 н. раствором KMnO4 до желтого цвета.
Титр раствора
 мг/мл, мг/мл,
V - 0,01 н. раствора KMnO4 пошедшего на титрование,
мл; 0,08 - титр 0,01 н. раствора KMnO4 по кислороду, мг/мл
|
|
Для получения раствора с титром 0,02 мг/мл его разбавляют дистиллатом
 мл; мл;
W - объем кислого индигокармина, мл
|
|
|
|
б) Определить кислотность раствора
|
б) 0,1 н. раствор NaOH
|
б) К 10 мл кислого раствора индигокармина (с титром 0,02 мг/мл О2)
прибавить 50 мл дистиллата и титровать 0,1 н. раствором щелочи с индикатором
фенолфталеином
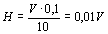
где V - 0,1 н.
раствора щелочи, пошедшего на титрование, мл
|
|
Индигокармин аммиачный
|
Щелочность 0,2 н.
|
Кислый раствор индигокармина
|
|
|
а) 5 н. раствор аммиака. Разбавить 37,5 мл 25 %-ного раствора NH4OH до 100 мл дистиллатом
|
|
|
|
б) Разбавление кислого раствора индигокармина 5 н. раствором аммиака
(по расчету)
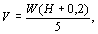
где W - объем взятого кислого
раствора индигокармина, мл; Н - кислотность кислого раствора
индигокармина, выраженная через нормальность; 0,2 - заданная нормальность
аммиачного раствора по аммиаку
|
0,1 н. раствор кислоты
|
Для проверки щелочности индигокарминового раствора к 5 мл аммиачного
раствора индигокармина прибавить 50 мл дистиллата, две капли смешанного
индигокармина и титровать 0,1 н. раствором кислоты до перехода синей окраски
в фиолетовую
|
|
Стандартный раствор фосфатов
|
100 мг/кг РО43-
|
Фосфат калия однозамещенный KH2РО4, х. ч.
|
Высушить В эксикаторе над серной кислотой в течение суток
|
0,1433 г
|
Навеску растворить в мерной литровой колбе и раствор довести до метки
дистиллатом
|
|
|
|
Стандартный раствор хлористого аммония
|
100 мг/кг NH3
|
Хлористый аммоний NH4Cl, х. ч.
|
Высушить при температуре 110 °С 1-2 ч
|
0,3147 г
|
Навеску растворить в Н-катионированной воде в мерной литровой колбе до
одного литра
|
|
|
|
10 мг/кг NH3
|
|
|
|
Стандартный раствор 100 мг/кг NH3 разбавить в 10 раз
|
|
|
|
Стандартный раствор железа
|
100
мг/кг Fe3+
|
Железоаммонийные квасцы NH4Fe х (SO4)2 х 12Н2O,
х. ч.
|
Высушить в эксикаторе при нормальной температуре
|
0,8634 г
|
Навеску растворить в мерной литровой колбе в дистиллате с добавлением
20 мл концентрированной серной кислоты, довести объем раствора до метки
|
|
|
|
10
мг/кг Fe3+
|
|
|
|
Стандартный раствор железа 100 мг/кг разбавить в 10 раз
|
|
|
|
|
